- MBTI
- .
- Enneagram
- 86_47
http://www.washingtonpost.com/blogs...alifornias-epidemic-of-vaccine-denial-mapped/California’s epidemic of vaccine denial, mapped
January 27The Disneyland measles outbreak has shined a harsh spotlight on anti-vaccine communities in California and around the country. Dozens of people have fallen ill with the disease, the vast majority of whom are unvaccinated. The year 2014 was already a banner year for measles in the U.S., with preliminary data from the Centers for Disease Control and Prevention showing the highest number of cases in over two decades.
My colleague Jason Millman reported last week on the results of a study showing that vaccine skeptics tend to cluster together in like-minded, often wealthy communities. Since the study focused on just a handful of northern California counties, I'd like to extend that analysis by looking at vaccine refusal across the entire state of California-- as a result both of vaccine denial and religious objections.
The map below is a first crack at that. Using data from the California Department of Public Health, it divides the state into equal-area grids, and within each area it counts the number of public school kindergartners with what's known as a "personal belief exemption" (PBE) to state vaccine requirements. The hexagon markers are sized by the total number of kindergartners in the area, and colored according to the percent of them with PBEs.

It's important to note a couple things. First, the actual number of kindergartners entering school without their full slate of vaccines is considerably higher than this map suggests. This map only represents kids whose parents have taken a stance against vaccines by applying for a PBE. Statewide about 2.5 percent of kindergartners fall into this category. Overall nearly 10 percent of California kindergartners don't start school fully vaccinated. Most of these kids are "conditional entrants," who, for whatever reasons, are behind in their vaccines but plan to get them.
It's also worth noting that the map only shows public school kids, as geographic data for private schools wasn't available. Kindergartners in private school are more than twice as likely (5.3 percent) to have a PBE than public school kids (2.5 percent). Still, the map provides a useful barometer of the level of principled opposition to vaccines in the state, and that's how I'll be discussing it.
Orange County, home to Disneyland, stands out as a cluster of dark red, as do parts of coastal San Diego county. The wealthy southern California coast from Malibu up to San Luis Obispo also stands out. Farther north, Santa Cruz, Marin and Sonoma counties are dark red. Inland there's a belt of anti-vaccine intensity from about Fresno, up through Sacramento and into Yuba county. Pockets of high PBE rates dot most of the northern California landscape.
On the other hand, the Los Angeles area is generally vaccine-friendly, as are much of the San Francisco and Silicon Valley regions. The relatively low-income areas in the central valley also have low rates of vaccine opposition.
Since California provides district-level vaccination data going back to 2000, it's also possible to map the spread of anti-vaccine sentiment over time. Check out the three maps below.
Back in 2000, only 0.77 percent of California kindergartners had personal belief exemptions from vaccines. By 2013, that percentage had more than quadrupled to 3.15 percent.
The scale on these maps tops off at 5 percent. But in some individual school districts, the actual PBE rate is much, much higher. At River Springs Charter School in Temecula, California, nearly a quarter of the 556 kindergartners had personal belief exemptions this year. A third of the kindergartners at the Visions in Education public school in Carmichael hold PBEs, as do 51 percent of kindergartners at Ocean Grove Charter School in Boulder Creek. At a handful of private schools, the PBE rate is 75 percent or more.
It goes without saying that a measles outbreak in these communities could be devastating. These parents are gambling with the lives not only of their children, but of other kids in their communities as well -- especially of young children and toddlers who aren't old enough to get all their vaccines yet. According to USA Today, six of the Disneyland measles cases were among infants too young to be vaccinated.
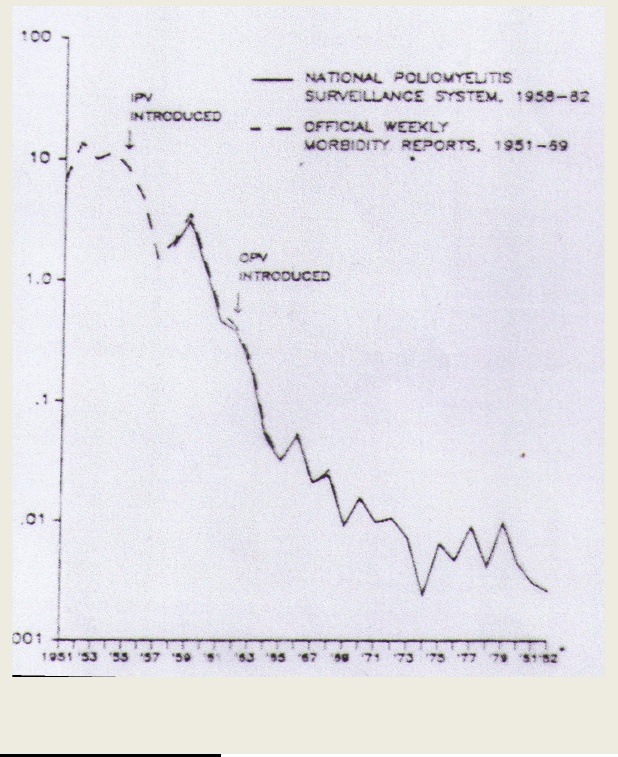
We need what's known as "herd immunity" to keep these most vulnerable populations safe from infectious diseases. For measles, that immunity kicks in with vaccination rates somewhere between 83 and 94 percent, according to the CDC. So even letting five percent of the kids in your community skip the vaccine is inviting trouble.
Fortunately, the news out of California isn't all bad. Starting with the current school year, California parents are required to consult a health care professional before being certified for a PBE. That's resulted in a drop in the PBE rate from 3.2 percent last year to 2.5 percent this year -- a welcome trend. The Disneyland outbreak also seems to be giving the issue enough visibility that some Californians previously on the fence are willing to change their minds.
I'm also singling out California simply because it does such a great job of making these numbers publicly available -- as far as the states go, it's far from the bottom of the pack on vaccination rates. According to numbers compiled by the Bloomberg Data team, California is ranked 39th in the nation on overall vaccination rates, with 65.3 percent of its children receiving all seven CDC-recommended shots. By contrast the bottom-ranked state, Alaska, only has a 59.1 percent vaccination rate, while the top, Mississippi, has a rate of 76 percent.
With any luck, in the aftermath of the California measles outbreak we'll see these number climb.